Hemophilia: from disease of kings to orphan disease
Share
Hemophilia includes a group of X-linked inherited diseases caused by mutations in the genes for coagulation factors FVIII (hemophilia A) and FIX (hemophilia B) and associated with bleeding. Affected patients may have mild, moderate or severe forms of the disease, depending on the plasma level of each factor. Despite being well recognized by the general public, it is considered an orphan disease, with a prevalence of 1 in 5,000 live male births in the case of hemophilia A and 1 in 30,000 in the case of hemophilia B. (1).
The peculiarities of hemophilia have been recognized since ancient times. The Talmud indicated that male infants should not be circumcised if two brothers had died from bleeding associated with the procedure. A 12th century description by the Arab physician Albucasis describes a family whose male members die from bleeding after minor injuries. In modern times, the physician John Conrad Otto, of Philadelphia-USA, published in 1803 the "Report of the hemorrhagic disposition existing in certain families."However, the term "hemophilia" was first used in 1828, in a paper by Hopff of the University of Zurich. Hemophilia has been called "the disease of kings", since numerous members of European royal houses of the time suffered from it, because Queen Victoria of England was a carrier of hemophilia B, as were her daughters Alice and Beatrice, who transmitted the disease to the Spanish, Russian and German royal houses; in addition, her son Leopold suffered from frequent hemorrhages and died at age 31 from a cerebral hemorrhage. (2).
Initially, it was thought that the hemorrhagic tendency of hemophilia was due to fragility of the blood vessels; in the 1930s, it was proposed that it was associated with alterations in the platelets. Subsequently, in 1937, it was found that by adding a substance, which at that time was called "antihemophilic globulin", to the patient's blood, the clotting defect was corrected. In 1944, Pavlosky's observations showed that the blood of one hemophilia patient corrected the clotting defect of another patient, and vice versa, demonstrating the defect in two distinct clotting factors, FVIII and FIX (3). These findings allowed an accurate diagnosis and the development of an appropriate treatment for this disease.
Etiology and pathophysiology
To maintain hemostasis, the human body has a complex system that allows the balance of procoagulant, anticoagulant and fibrinolytic states to maintain blood fluidity within the vascular system and to achieve rapid thrombus development in response to vascular injury. In this process, the sequential activation of complexes composed of vitamin K-dependent factors (FVII, FIX, FX) and their respective cofactors (tissue factor, FVIII, FV) is essential. (4). FVIII and FIX factors are part of the intrinsic coagulation pathway (see figure 1).
Clinical features
The clinical presentation of hemophilia A and B is similar, with the joints being the main site of spontaneous bleeding, mainly the ankle, followed by the elbows and knees. Approximately half of the children with severe hemophilia develop intramuscular hematomas as early as 6 to 8 months of age (5).
Patients with mild hemophilia usually bleed excessively only in association with surgery or major trauma; however, those with severe hemophilia have frequent episodes of spontaneous bleeding, especially intra-articular or muscular, following minor trauma. (6).
Recurrent joint bleeding induces a cascade of inflammatory and degenerative processes that injure the synovium, cartilage and bone. The major trigger for these processes is iron released into the synovial fluid, which has proinflammatory and angiogenic activity; the associated neovascularization leads to the formation of new friable vessels, more prone to bleeding, leading to a cycle of bleeding, iron accumulation, synovial hypertrophy, and rebleeding (7).
Due to these cycles, many patients develop chronic synovitis with joint edema; others develop hemophilic arthropathy with severe osteochondral damage. This arthropathy leads to chronic pain, loss of range of motion, muscle atrophy, and, because of this, reduced quality of life (8). Another frequent complication is intracranial hemorrhage, with an incidence of 1.9% and a mortality rate of 19.6% (9). (9).

Diagnosis and follow-up
The diagnosis of hemophilia is relatively simple. Upon suspicion, the activity of coagulation factors FVIII for hemophilia A and FIX for hemophilia B should be measured. For this, it should be taken into account that the levels of vitamin K-dependent coagulation factors, including factor IX, are decreased at birth, so hemophilia B can be difficult to diagnose in neonates and the measurement should be repeated at 6 months if the diagnostic suspicion persists. On the other hand, FVIII deficiency or hemophilia A can be diagnosed at birth, even with a cord blood sample ( 10).
According to the residual activity of the affected factor, hemophilia is classified as mild, moderate or severe (11); the age of symptom onset, the type of symptoms, the rate of spontaneous intra-articular bleeding and the development of inhibitors depend on this (see Table 1).
The identification of the genetic mutation present is essential, since it can predict the risk of inhibitor formation (12), as well as identify female carriers of the disease in the family. Between 40% and 45% of patients with severe hemophilia A have inversion of intron 22 of the F8 gene, while 1% to 6% have inversion of intron 1. In cases of mild and moderate hemophilia A, complete sequencing of the F8 gene should be performed. In hemophilia B, the sequence of 8 exons, the intron-exon boundaries and the promoter region of F9 are analyzed (13).
In the follow-up of patients it is essential to assess the presence of inhibitors. The development of inhibitors is, at present, the most difficult complication derived from treatment with coagulation factors. These are alloantibodies that are present in up to 20% to 30% of patients with hemophilia A and 1% to 5% of those with hemophilia B (14), which makes the infusion of concentrated FVIII inefficient, increases patient morbidity and mortality and reduces their quality of life.
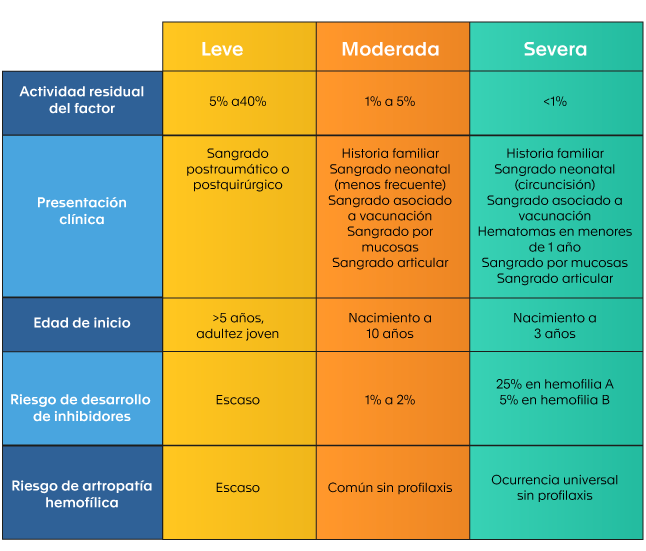
Table 1. Classification of hemophilia according to clinical presentation (4).
The formation of these inhibitors is a complex and multifactorial process; it has been suggested in some studies that it may vary according to the type of product used in the treatment (plasma-derived versus recombinant FVIII); however, the evidence in this regard is inconclusive.
Inhibitors are high-affinity polyclonal IgG antibodies directed against the FVIII protein and can be of two types: type 1, which completely inactivate FVIII, and type 2, which incompletely inactivate FVIII. Patients with high antibody titers develop severe hemorrhagic complications that do not respond to clotting factor replacement (15). Although many of these inhibitors are transient, or resolve with immunotherapy, up to 15% of patients with hemophilia A and 3% of those with hemophilia B have persistent antibodies. (16).
Treatment: historical evolution
In the 1950s and 1960s, patients with hemophilia were treated with fresh blood or plasma only; however, because the low concentration of clotting factors required in these products was not sufficient to treat severe bleeds, a large number of patients died in infancy or young adulthood. (17).
In 1964, it was discovered that the cryoprecipitated fraction of plasma contains large amounts of FVIII. This made it possible to concentrate sufficient amounts of the factor in smaller volumes and to perform larger surgeries. However, the modern era of hemophilia treatment began in the 1970s with the production of lyophilized clotting factor concentrates. This advance made it possible to improve the quality and prolong the life expectancy of patients due to the implementation and dissemination of home replacement therapy, early control of hemorrhages, and the reduction of musculoskeletal damage associated with inadequate treatment (2).
Primary prophylaxis was able to prevent most episodes of spontaneous joint bleeding, reducing arthropathy. On the other hand, the discovery of desmopressin, which shortens the prolonged activated partial thromboplastin time and bleeding time, favoring the elevation of FVIII and von Willebrand factor, has become an economical and safe treatment for patients with mild hemophilia A (18).
Unfortunately, the era of plasma concentrates for the treatment of hemophilia suffered a major setback due to the transmission of human immunodeficiency virus (HIV) and Hepatitis C virus (HCV) through clotting factor concentrates made from plasma from multiple donors (17). Thousands of hemophilia patients died from complications of HIV/AIDS in the 1980s and 1990s. As a consequence, safer treatments were sought and viral inactivation techniques were implemented in the production of plasma-derived factor concentrates and viral screening in donated blood, all of which increased the safety of these derivatives (17).

The greatest advance in the treatment of hemophilia occurred in the last decades of the 20th century and was related to the progress of recombinant DNA technology, which allowed the development of recombinant FVIII and FIX, thus reducing the risk of pathogen transmission (19). (19). As a result of progress in therapy for the hemophilias, the life expectancy of patients has caught up with that of the general population. This has led to the development of age-associated chronic diseases, previously unseen in the hemophilia population (1).
Recent therapeutic approaches include specific antibodies that simulate the coagulant function of FVIII (20)(20), inhibition of anticoagulant proteins, such as antithrombin, with RNA interference molecules, and the tissue factor inhibitory pathway with monoclonal antibodies (21)(22).
On the other hand, phase 3 studies of gene therapy are underway as a curative option against hemophilia. The goal of gene therapy is that patients do not require coagulation factor replacement therapy and do not bleed. Successful gene therapy results in endogenous expression of clotting factor, reaching a stable level and with a sustained duration of action. This would make prophylaxis and intravenous infusions unnecessary. In addition to that, it has been postulated that endogenous expression of the factors could be less immunogenic, decreasing or avoiding the generation of inhibitors (23).
In the future, it is expected to have an increase in the production of FVIII and FIX concentrates to supply developing countries, in addition to the processing of molecules with longer half-life and lower immunogenicity. (2).
World Hemophilia Day
The World Federation of Hemophilia (WFH), since 1989, commemorates every year on April 17 World Hemophilia Day, in honor of the birthday of its founder, Frank Schnabel (24). The objective of this day is to bring the community with bleeding disorders closer together and unite them. For this purpose, hemophilia patient organizations and specialized treatment centers carry out academic and sports activities, exhibitions and other events to raise awareness in the community about hemophilia, its complications and implications in the lives of patients who suffer from it.
In 2021, the WFH took as its theme for this day "ADAPTING TO CHANGE: PRESERVING CARE IN A NEW WORLD"because of the severe impact the COVID-19 pandemic has had on people with bleeding disorders and the global changes it has brought. The WFH persists in its goal of ensuring access for all people with such disorders to appropriate and sustainable treatment and care, adapting to the changes made necessary by the pandemic (25).
Hemophilia in Colombia
In the country, hemophilia, being in the census of orphan diseases, is listed as mandatory reporting in the National Public Health Surveillance System - Sivigila. The Colombian League of Hemophiliacs intervenes, through activism and representation before scientific societies, so that decisions and national management guidelines take into account the integral benefit of patients (26).
By 2017 in Colombia, 2170 people with hemophilia were registered, of whom 1794 had hemophilia A and 376 had hemophilia B, and 1167 were classified as severe hemophilia. Of these patients, 1284 were on prophylaxis and 324 (14.9% of the total) had inhibitors. During the same period, 16 patients with hemophilia died; however, only two of them died due to complications associated with the disease (27).
Recently, the National Registry of Hemophilia and other coagulopathies was published as a multisectoral initiative to centralize sociodemographic, clinical and economic information on these patients, by means of a consensus of experts, in order to monitor morbidity and mortality, evaluate access to health services, their impact on the complications of the disease and the costs associated with medical care. This registry is expected to guide rational decision making for an efficient use of economic resources, as well as to promote health research to improve the quality of life and reduce the associated disabilities in patients with hemophilia (28).
In the Laboratorio Clínico Hematológico
The Laboratorio Clínico Hematológico offers tests for the evaluation of hemostasis divided into two areas following the physiological order: platelet aggregation and coagulation studies specific to primary hemostasis and studies associated with factors and dynamics of secondary hemostasis. These tests are essential in the study of patients with abnormal bleeding conditions such as hemophilia or von Willebrand's disease. Within the specialized tests for the diagnosis of bleeding disorders, we offer the determination of coagulation factors VIII, IX and XI, for the diagnosis of deficiencies and as a follow-up of factor VIII and IX replacement therapy.
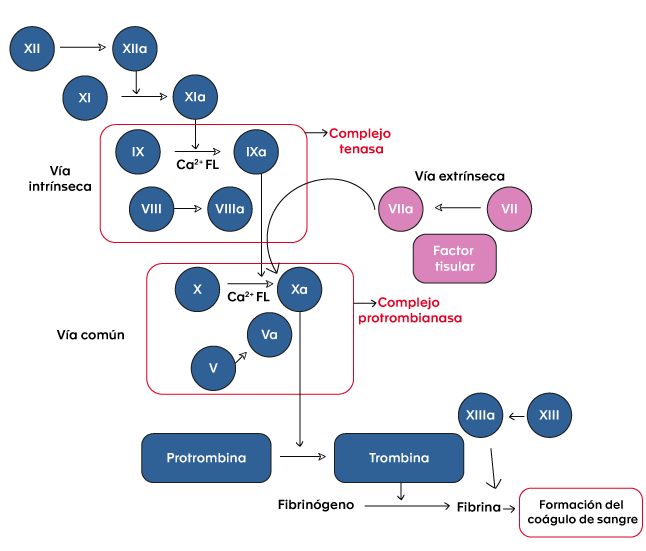
Figure 1. Coagulation cascade.
Bibliography
- Franchini M. The modern treatment of haemophilia: a narrative review. Blood Transfus. April 2013;11(2):178-82.
- Franchini M, Mannucci P. Past, present and future of hemophilia: a narrative review. Orphanet J Rare Dis. 2012;7(1):24.
- Biggs R. CHRISTMAS DISEASE. A CONDITION PREVIOUSLY MISTAKEN FOR HAEMOPHILIA. British Medical Journal. December 27, 1952;1378-82.
- Kizilocak H, Young G. Diagnosis and Treatment of Hemophilia. Clin Adv Hematol Oncol. 2019;17(6):8.
- Van Den Berg HM, De Groot PHG, Fischer K. Phenotypic heterogeneity in severe hemophilia: Prothrombotic risk factors and phenotype of severe hemophilia. J Thromb Haemost. July 9, 2007;5:151-6.
- Hoyer LW. Hemophilia A. N Engl J Med. Jan 6, 1994;330(1):38-47.
- Acharya SS, Kaplan RN, Macdonald D, Fabiyi OT, DiMichele D, Lyden D. Neoangiogenesis contributes to the development of hemophilic synovitis. Blood. Feb 24, 2011;117(8):2484-93.
- Jansen NWD, Roosendaal G, Lafeber FPJG. Understanding haemophilic arthropathy: an exploration of current open issues. Br J Haematol. 2008;143(5):632-40.
- Witmer C, Presley R, Kulkarni R, Michael Soucie J, Manno CS, Raffini L. Associations between intracranial haemorrhage and prescribed prophylaxis in a large cohort of haemophilia patients in the United States: Intracranial Haemorrhage in Haemophilia. Br J Haematol. Jan 2011;152(2):211-6.
- Attard C, van der Straaten T, Karlaftis V, Monagle P, Ignjatovic V. Developmental hemostasis: age-specific differences in the levels of hemostatic proteins. J Thromb Haemost. Oct 2013;11(10):1850-4.
- Blanchette VS, Key NS, Ljung LR, Manco-Johnson MJ, van den Berg HM, Srivastava A, et al. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. November 2014;12(11):1935-9.
- Gouw SC, van den Berg HM, Oldenburg J, Astermark J, de Groot PG, Margaglione M, et al. F8 gene mutation type and inhibitor development in patients with severe hemophilia A: systematic review and meta-analysis. Blood. Mar 22, 2012;119(12):2922-34.
- Bagnall RD, Waseem N, Green PM, Giannelli F. Recurrent inversion breaking intron 1 of the factor VIII gene is a frequent cause of severe hemophilia A. Blood. Jan 1, 2002;99(1):168-74.
- Peyvandi F, Garagiola I, Young G. The past and future of haemophilia: diagnosis, treatments, and its complications. The Lancet. July 2016;388(10040):187-97.
- Cannavò A, Valsecchi C, Garagiola I, Palla R, Mannucci PM, Rosendaal FR, et al. Nonneutralizing antibodies against factor VIII and risk of inhibitor development in severe hemophilia A. Blood. Mar 9, 2017;129(10):1245-50.
- Gringeri A, Mantovani LG, Scalone L, Mannucci PM, the COCIS Study Group. Cost of care and quality of life for patients with hemophilia complicated by inhibitors: the COCIS Study Group. Blood. Oct 1, 2003;102(7):2358-63.
- Mannucci PM. Back to the future: a recent history of haemophilia treatment. Haemophilia. July 2008;14(s3):10-8.
- Mannucci PM, Tuddenham EGD. The Hemophilias - From Royal Genes to Gene Therapy. N Engl J Med. 2001;7.
- Pipe S. Recombinant clotting factors. Thromb Haemost. 2008;99(11):840-50.
- Shima M, Hanabusa H, Taki M, Matsushita T, Sato T, Fukutake K, et al. Factor VIII-Mimetic Function of Humanized Bispecific Antibody in Hemophilia A. N Engl J Med. May 26, 2016;374(21):2044-53.
- Petersen LC. Hemostatic properties of a TFPI antibody. Thromb Res. May 2012;129:S44-5.
- Shapiro AD, Angchaisuksiri P, Astermark J, Benson G, Castaman G, Chowdary P, et al. Subcutaneous concizumab prophylaxis in hemophilia A and hemophilia A/B with inhibitors: phase 2 trial results. Blood. Nov 28, 2019;134(22):1973-82.
- Pipe SW. Gene therapy for hemophilia. Pediatr Blood Cancer. February 2018;65(2):e26865.
- April 17 - World Hemophilia Day > Pfizer.com [Internet]. [cited 2021 Mar 31, 2021]. Available from: https://www.pfizer.es/salud/dias_salud/17_abril_dia_mundial_hemofilia.html
- World hemophilia day 2021 - World Federation of Hemophilia [Internet]. [cited Mar 31, 2021]. Available from: https://www.wfh.org/es/eventos/dia-world-haemophilia-day .
- Colombian League of Hemophiliacs and other blood deficiencies [Internet]. [cited Mar 2, 2021]. Available from: http://colhemofilicos.org.co/
- Acuña L. Hemophilia situation in Colombia 2017.pdf [Internet]. Bogotá; 2018. 176 p. Available at: http://colhemofilicos.org.co/_assets/archives/presentaciones/Libro_situacion_hemofilia_en%20Colombia_2017.pdf
- Alvis LF, Sánchez P, Acuña L, Escobar G, Linares A, Solano MH, et al. National registry of haemophilia and other coagulopathies: A multisector initiative in the Colombian Health System. Haemophilia [Internet]. November 2020 [cited March 24, 2021];26(6). Available from: https://onlinelibrary.wiley.com/doi/10.1111/hae.14138
{kind=link}