Hemofilia: de enfermedad de reyes a enfermedad huérfana
Compartir
La hemofilia incluye un grupo de enfermedades hereditarias ligadas al cromosoma X, causadas por mutaciones en los genes de los factores FVIII (hemofilia A) y FIX (hemofilia B) de la coagulación, y asociadas a sangrado. Los pacientes afectados pueden presentar formas leves, moderadas o severas de la enfermedad, según el nivel plasmático de cada factor. A pesar de ser muy reconocida por el público general, se considera una enfermedad huérfana, ya que su prevalencia es de 1 en 5.000 nacidos vivos de sexo masculino en el caso de la hemofilia A, y de 1 en 30.000 en el caso de la hemofilia B (1).
Desde tiempos antiguos se han reconocido las particularidades de la Hemofilia. El Talmud indicaba que los bebés de sexo masculino no debían ser circuncidados si dos hermanos habían muerto por sangrado asociado al procedimiento. Una descripción del siglo XII, del médico árabe Albucasis describe una familia cuyos miembros masculinos fallecen por sangrado luego de lesiones menores. En tiempos modernos, el médico John Conrad Otto, de Filadelfia-Estados Unidos, publicó en 1803 el “Reporte de la disposición hemorrágica existente en ciertas familias”; sin embargo, el término “hemofilia” fue usado por primera vez en 1828, en un escrito de Hopff de la Universidad de Zurich. La hemofilia ha sido llamada “la enfermedad de reyes”, ya que numerosos miembros de casas reales europeas de la época la sufrían, debido a que la reina Victoria de Inglaterra era portadora de hemofilia B, al igual que sus hijas Alicia y Beatriz, que transmitieron la enfermedad a las casas reales española, rusa y alemana; además, su hijo Leopoldo sufría de hemorragias frecuentes y falleció a los 31 años de una hemorragia cerebral (2).
En un principio, se pensaba que la tendencia hemorrágica de la hemofilia se debía a fragilidad de los vasos sanguíneos; en los años 30, se propuso que estaba asociada a alteraciones en las plaquetas. Posteriormente, en 1937, se encontró que al adicionar una sustancia, que en ese momento fue llamada “globulina antihemofílica”, a la sangre del paciente, se corregía el defecto de la coagulación. En 1944, las observaciones de Pavlosky demostraron que la sangre de un paciente con hemofilia corregía el defecto de la coagulación de otro paciente, y viceversa, demostrando el defecto en dos factores distintos de la coagulación, FVIII y FIX (3). Estos hallazgos permitieron un diagnóstico preciso y el desarrollo de un tratamiento apropiado para esta enfermedad.
Etiología y fisiopatología
Para mantener la hemostasia, el cuerpo humano tiene un sistema complejo que permite el balance de los estados procoagulante, anticoagulante y fibrinolítico, para mantener la fluidez de la sangre dentro del sistema vascular y lograr desarrollar un trombo rápidamente como respuesta a lesiones vasculares. En este proceso, es esencial la activación secuencial de los complejos compuestos por factores dependientes de vitamina K (FVII, FIX, FX) y sus respectivos cofactores (factor tisular, FVIII, FV) (4). Los factores FVIII y FIX hacen parte de la vía intrínseca de la coagulación (ver figura 1).
Características clínicas
La presentación clínica de la hemofilia A y B es similar, siendo las articulaciones el principal sitio de sangrado espontáneo, principalmente el tobillo, seguido de los codos y las rodillas. Aproximadamente la mitad de los niños con hemofilia severa desarrollan hematomas intramusculares desde los 6 a 8 meses de edad (5).
Los pacientes con hemofilia leve suelen sangrar excesivamente únicamente en asociación a cirugía o trauma mayor; sin embargo, aquellos con hemofilia severa presentan episodios frecuentes de sangrado espontáneo, especialmente intraarticular o muscular, luego de traumas menores (6).
El sangrado articular recurrente induce una cascada de procesos inflamatorios y degenerativos que lesionan la sinovia, el cartílago y el hueso. El mayor detonante de estos procesos es el hierro liberado al líquido sinovial, que tiene actividad proinflamatoria y angiogénica; la neovascularización asociada lleva a la formación de nuevos vasos friables, más propensos al sangrado, lo que lleva a un ciclo de sangrado, acumulación de hierro, hipertrofia sinovial y nuevo sangrado (7).
Debido a estos ciclos, muchos pacientes desarrollan sinovitis crónica con edema articular; otros llegan a la artropatía hemofílica con daño osteocondral severo. Esta artropatía lleva a dolor crónico, pérdida del rango de movimientos, atrofia muscular y, debido a esto, reducción de la calidad de vida (8). Otra complicación frecuente es la hemorragia intracraneana, con una incidencia del 1.9% y una tasa de mortalidad del 19.6% (9).

Diagnóstico y seguimiento
El diagnóstico de la hemofilia es relativamente sencillo. Ante la sospecha, se debe realizar medición de la actividad de los factores de la coagulación FVIII para la hemofilia A y FIX para la hemofilia B. Para esto, se debe tener en cuenta que los niveles de los factores de coagulación dependientes de vitamina K, entre ellos el factor IX, están disminuidos al nacer, por lo que la hemofilia B puede ser difícil de diagnosticar en neonatos y la medición se debe repetir a los 6 meses si persiste la sospecha diagnóstica. Por otro lado, la deficiencia de FVIII o hemofilia A se puede diagnosticar al nacer, incluso con una muestra de sangre del cordón umbilical (10).
Según la actividad residual del factor afectado, la hemofilia se clasifica en leve, moderada o severa (11); de esto depende la edad de inicio de los síntomas, el tipo de síntomas, la tasa de sangrados intraarticulares espontáneos y el desarrollo de inhibidores (ver Tabla 1).
La identificación de la mutación genética presente es esencial, ya que puede pronosticar el riesgo de formación de inhibidores (12), así como identificar mujeres portadoras de la enfermedad en la familia. Del 40% al 45% de los pacientes con hemofilia A severa tienen inversión del intrón 22 del gen F8, mientras que 1% al 6%, inversión del intrón 1. Al ser estas las mutaciones más comunes, se hace búsqueda inicial de éstas, con posterior secuenciación completa del gen en caso de que no se encuentren. En casos de hemofilia A leve y moderada, se debe hacer secuenciación completa del gen F8. En la hemofilia B, se analiza la secuencia de 8 exones, los límites intrón-exón y la región promotora de F9 (13).
En el seguimiento de los pacientes es esencial valorar la presencia de inhibidores. El desarrollo de inhibidores es, en la actualidad, la complicación más difícil derivada del tratamiento con factores de coagulación. Estos son aloanticuerpos que se presentan hasta en 20% al 30% de los pacientes con hemofilia A y 1% al 5% de aquellos con hemofilia B (14), lo que hace poco eficiente la infusión de FVIII concentrado, aumenta la morbimortalidad de los pacientes y disminuye su calidad de vida.
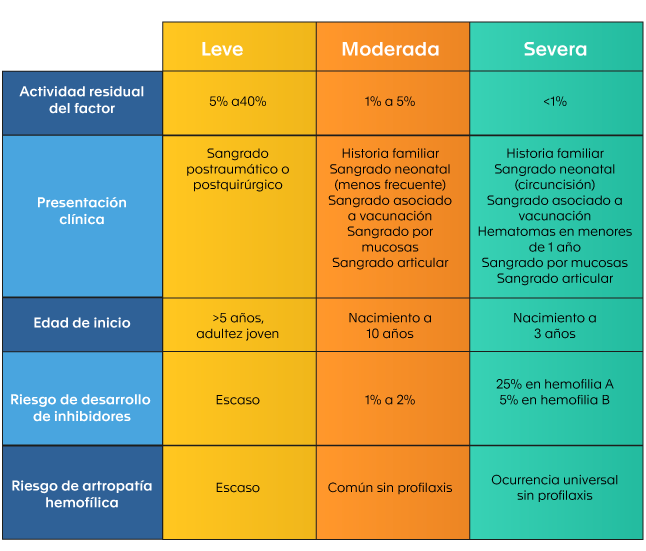
Tabla 1. Clasificación de la hemofilia según presentación clínica (4).
La formación de estos inhibidores es un proceso complejo y multifactorial; se ha sugerido en algunos estudios que puede variar según el tipo de producto utilizado en el tratamiento (FVIII derivado del plasma versus FVIII recombinante); sin embargo, la evidencia al respecto no es concluyente.
Los inhibidores son anticuerpos IgG policlonales de alta afinidad dirigidos contra la proteína del FVIII y pueden ser de dos tipos: los tipo 1, que inactivan completamente el FVIII, y los tipo 2, que lo hacen de manera incompleta. Los pacientes con altos títulos de anticuerpos desarrollan complicaciones hemorrágicas severas que no responden al reemplazo de factores de coagulación (15). Aunque muchos de estos inhibidores son transitorios, o se resuelven con inmunoterapia, hasta el 15% de los pacientes con Hemofilia A y el 3% de aquellos con hemofilia B presentan anticuerpos persistentes (16).
Tratamiento: evolución histórica
En los años 50 y 60, los pacientes con hemofilia eran tratados con sangre o plasma fresco únicamente; sin embargo, debido a que la baja concentración de los factores de coagulación requeridos en estos productos no era suficiente para tratar sangrados severos, un gran número de pacientes moría en la infancia o adultez joven (17).
En 1964, se descubrió que la fracción crioprecipitada del plasma contiene grandes cantidades de FVIII. Esto permitió concentrar suficiente cantidad del factor en volúmenes menores y la realización de cirugías mayores. Sin embargo, la era moderna del tratamiento de la hemofilia inició en los años 70 con la producción de concentrados liofilizados de factores de coagulación. Este avance permitió mejorar la calidad y prolongar la expectativa de vida de los pacientes debido a la implementación y difusión de la terapia de reemplazo en casa, el control temprano de hemorragias y la disminución del daño músculo-esquelético asociado al tratamiento inadecuado (2).
La profilaxis primaria logró prevenir la mayoría de los episodios de sangrado articular espontáneo, disminuyendo la artropatía. Por otro lado, el descubrimiento de la desmopresina, que acorta el tiempo de tromboplastina parcial activada prolongado y el tiempo de sangrado, favoreciendo la elevación del FVIII y del factor de von Willebrand, ha logrado ser un tratamiento económico y seguro para los pacientes con hemofilia A leve (18).
Desafortunadamente, la era de los concentrados de plasma para el tratamiento de las hemofilias sufrió un gran revés, debido a la transmisión del virus de la inmunodeficiencia humana (VIH) y de la Hepatitis C (VHC), a través de los concentrados de factores de coagulación fabricados a partir de plasma de múltiples donantes (17). Miles de pacientes con hemofilia fallecieron por complicaciones del VIH/SIDA en los 80 y 90. Como consecuencia de esto, se buscaron tratamientos más seguros y se implementaron técnicas de inactivación viral en la producción de los factores concentrados derivados del plasma y de tamización viral en sangre donada; todo esto aumentó la seguridad de dichos derivados (17).

El mayor avance en el tratamiento de las hemofilias se dio en las últimas décadas del siglo XX y tuvo que ver con el progreso de la tecnología de ADN recombinante, lo que permitió el desarrollo de FVIII y FIX recombinantes y disminuyó, así, el riesgo de transmisión de patógenos (19). Como resultado del progreso en la terapia para las hemofilias, la esperanza de vida de los pacientes se ha equiparado a la de la población general. Esto ha llevado al desarrollo de enfermedades crónicas asociadas a la edad, previamente no vistas en la población con hemofilia (1).
Entre las aproximaciones terapéuticas recientes, se encuentran anticuerpos específicos que simulan la función coagulante del FVIII (20), inhibición de proteínas anticoagulantes, como la antitrombina, con moléculas de interferencia del ARN, y la vía inhibidora del factor tisular con anticuerpos monoclonales (21)(22).
Por otro lado, se encuentran en curso estudios fase 3 de terapia génica, como opción curativa contra las hemofilias. La meta de la terapia génica es que los pacientes no requieran terapia de reemplazo de factores de coagulación y no presenten sangrados. La terapia génica exitosa resulta en la expresión endógena del factor de coagulación, llegando a un nivel estable y con una duración de acción sostenida. Esto haría innecesarias la profilaxis y las infusiones intravenosas. Además de eso, se ha postulado que la expresión endógena de los factores podría ser menos inmunogénica, disminuyendo o evitando la generación de inhibidores (23).
A futuro, se espera contar con incremento en la producción de concentrados de FVIII y FIX para suplir a países en desarrollo; además, del procesamiento de moléculas con mayor vida media y menor inmunogenicidad (2).
Día Mundial de la Hemofilia
La Federación Mundial de la Hemofilia (FMH), desde 1989, conmemora cada año el 17 de abril el Día Mundial de la Hemofilia, en honor al natalicio de su fundador, Frank Schnabel (24). El objetivo de este día es acercar y unir a la comunidad con trastornos de la coagulación, para esto, las organizaciones de pacientes con hemofilia y los centros especializados de tratamiento realizan actividades académicas, deportivas, exposiciones y otros eventos para sensibilizar a la comunidad sobre la hemofilia, sus complicaciones e implicaciones en la vida de los pacientes que la padecen.
En 2021, la FMH tomó como lema para este día “ADAPTARSE AL CAMBIO: PRESERVAR LA ATENCIÓN EN UN MUNDO NUEVO”, debido al fuerte impacto que la pandemia de COVID-19 ha tenido en las personas con trastornos de la coagulación y los cambios globales que ha implicado. La FMH persiste en su objetivo de asegurar el acceso de todas las personas con dichos trastornos a tratamientos y cuidados adecuados y sostenibles, adaptándose a los cambios que la pandemia ha hecho necesarios (25).
Hemofilia en Colombia
En el país, las Hemofilias, al estar en el censo de enfermedades huérfanas, está listada como de reporte obligatorio en el Sistema Nacional de Vigilancia en Salud Pública – Sivigila. La Liga Colombiana de Hemofílicos interviene, mediante el activismo y representación ante las sociedades científicas, para que las decisiones y las guías nacionales de manejo tengan en cuenta el beneficio integral de los pacientes (26).
Para el 2017 en Colombia estaban registrados 2170 personas con hemofilia, de los cuales 1794 presentaban hemofilia A y 376 hemofilia B, y 1167 clasificados como hemofilia severa. De estos pacientes, 1284 se encontraban en esquema de profilaxis y 324 (14.9% del total) tuvieron presencia de inhibidores. En ese mismo período, los pacientes con hemofilia fallecidos fueron 16; sin embargo, únicamente dos de ellos por complicaciones asociadas a la enfermedad (27).
Recientemente, se publicó el Registro Nacional de Hemofilia y otras coagulopatías, como una iniciativa multisectorial para centralizar la información sociodemográfica, clínica y económica de estos pacientes, por medio de un consenso de expertos, con objeto de monitorizar la morbilidad y mortalidad, evaluar el acceso a los servicios de salud, su impacto en las complicaciones de la enfermedad y los costos asociados a la asistencia médica. Se espera con este registro guiar la toma de decisiones racionales para un uso eficiente de los recursos económicos; además, de impulsar la investigación en salud para mejorar la calidad de vida y disminuir las discapacidades asociadas en los pacientes con hemofilias (28).
En el Laboratorio Clínico Hematológico
El Laboratorio Clínico Hematológico ofrece pruebas para la evaluación de la hemostasia dividida en dos áreas siguiendo el orden fisiológico: estudios de agregación plaquetaria y coagulación específicos a la hemostasia primaria y estudios asociados a factores y la dinámica de la hemostasia secundaria. Estas pruebas son esenciales en el estudio de pacientes con condiciones de sangrado anormal como hemofilia o enfermedad de von Willebrand. Dentro de las pruebas especializadas para el diagnóstico de desórdenes hemorrágicos, ofrecemos la determinación de los factores VIII, IX y XI de la coagulación, para el diagnóstico de deficiencias y como seguimiento de la terapia de sustitución de factores VIII y IX.
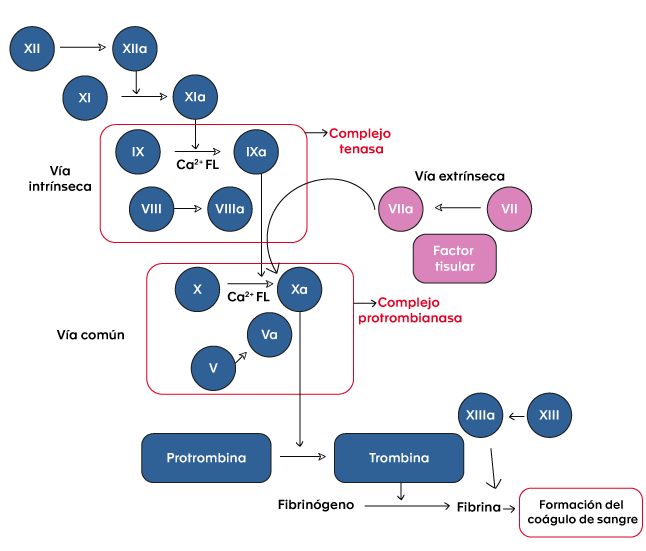
Figura 1. Cascada de la coagulación.
Bibliografía
- Franchini M. The modern treatment of haemophilia: a narrative review. Blood Transfus. abril de 2013;11(2):178-82.
- Franchini M, Mannucci P. Past, present and future of hemophilia: a narrative review. Orphanet J Rare Dis. 2012;7(1):24.
- Biggs R. CHRISTMAS DISEASE. A CONDITION PREVIOUSLY MISTAKEN FOR HAEMOPHILIA. British Medical Journal. 27 de diciembre de 1952;1378-82.
- Kizilocak H, Young G. Diagnosis and Treatment of Hemophilia. Clin Adv Hematol Oncol. 2019;17(6):8.
- Van Den Berg HM, De Groot PHG, Fischer K. Phenotypic heterogeneity in severe hemophilia: Prothrombotic risk factors and phenotype of severe hemophilia. J Thromb Haemost. 9 de julio de 2007;5:151-6.
- Hoyer LW. Hemophilia A. N Engl J Med. 6 de enero de 1994;330(1):38-47.
- Acharya SS, Kaplan RN, Macdonald D, Fabiyi OT, DiMichele D, Lyden D. Neoangiogenesis contributes to the development of hemophilic synovitis. Blood. 24 de febrero de 2011;117(8):2484-93.
- Jansen NWD, Roosendaal G, Lafeber FPJG. Understanding haemophilic arthropathy: an exploration of current open issues. Br J Haematol. 2008;143(5):632-40.
- Witmer C, Presley R, Kulkarni R, Michael Soucie J, Manno CS, Raffini L. Associations between intracranial haemorrhage and prescribed prophylaxis in a large cohort of haemophilia patients in the United States: Intracranial Haemorrhage in Haemophilia. Br J Haematol. enero de 2011;152(2):211-6.
- Attard C, van der Straaten T, Karlaftis V, Monagle P, Ignjatovic V. Developmental hemostasis: age-specific differences in the levels of hemostatic proteins. J Thromb Haemost. octubre de 2013;11(10):1850-4.
- Blanchette VS, Key NS, Ljung LR, Manco-Johnson MJ, van den Berg HM, Srivastava A, et al. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. noviembre de 2014;12(11):1935-9.
- Gouw SC, van den Berg HM, Oldenburg J, Astermark J, de Groot PG, Margaglione M, et al. F8 gene mutation type and inhibitor development in patients with severe hemophilia A: systematic review and meta-analysis. Blood. 22 de marzo de 2012;119(12):2922-34.
- Bagnall RD, Waseem N, Green PM, Giannelli F. Recurrent inversion breaking intron 1 of the factor VIII gene is a frequent cause of severe hemophilia A. Blood. 1 de enero de 2002;99(1):168-74.
- Peyvandi F, Garagiola I, Young G. The past and future of haemophilia: diagnosis, treatments, and its complications. The Lancet. julio de 2016;388(10040):187-97.
- Cannavò A, Valsecchi C, Garagiola I, Palla R, Mannucci PM, Rosendaal FR, et al. Nonneutralizing antibodies against factor VIII and risk of inhibitor development in severe hemophilia A. Blood. 9 de marzo de 2017;129(10):1245-50.
- Gringeri A, Mantovani LG, Scalone L, Mannucci PM, the COCIS Study Group. Cost of care and quality of life for patients with hemophilia complicated by inhibitors: the COCIS Study Group. Blood. 1 de octubre de 2003;102(7):2358-63.
- Mannucci PM. Back to the future: a recent history of haemophilia treatment. Haemophilia. julio de 2008;14(s3):10-8.
- Mannucci PM, Tuddenham EGD. The Hemophilias — From Royal Genes to Gene Therapy. N Engl J Med. 2001;7.
- Pipe S. Recombinant clotting factors. Thromb Haemost. 2008;99(11):840-50.
- Shima M, Hanabusa H, Taki M, Matsushita T, Sato T, Fukutake K, et al. Factor VIII–Mimetic Function of Humanized Bispecific Antibody in Hemophilia A. N Engl J Med. 26 de mayo de 2016;374(21):2044-53.
- Petersen LC. Hemostatic properties of a TFPI antibody. Thromb Res. mayo de 2012;129:S44-5.
- Shapiro AD, Angchaisuksiri P, Astermark J, Benson G, Castaman G, Chowdary P, et al. Subcutaneous concizumab prophylaxis in hemophilia A and hemophilia A/B with inhibitors: phase 2 trial results. Blood. 28 de noviembre de 2019;134(22):1973-82.
- Pipe SW. Gene therapy for hemophilia. Pediatr Blood Cancer. febrero de 2018;65(2):e26865.
- 17 de abril – Día Mundial de la Hemofilia > Pfizer.es [Internet]. [citado 31 de marzo de 2021]. Disponible en: https://www.pfizer.es/salud/dias_salud/17_abril_dia_mundial_hemofilia.html
- Día mundial de la hemofilia 2021 – World Federation of Hemophilia [Internet]. [citado 31 de marzo de 2021]. Disponible en: https://www.wfh.org/es/eventos/dia-mundial-de-la-hemofilia
- Liga Colombiana de Hemofílicos y otras deficiencias sanguíneas [Internet]. [citado 2 de marzo de 2021]. Disponible en: http://colhemofilicos.org.co/
- Acuña L. Situación de la hemofilia en Colombia 2017.pdf [Internet]. Bogotá; 2018. 176 p. Disponible en: http://colhemofilicos.org.co/_assets/archives/presentaciones/Libro_situacion_hemofilia_en%20Colombia_2017.pdf
- Alvis LF, Sánchez P, Acuña L, Escobar G, Linares A, Solano MH, et al. National registry of haemophilia and other coagulopathies: A multisector initiative in the Colombian Health System. Haemophilia [Internet]. noviembre de 2020 [citado 24 de marzo de 2021];26(6). Disponible en: https://onlinelibrary.wiley.com/doi/10.1111/hae.14138
{kind=link}